Exploring organic, inorganic, main-group, and materials reactivity through molecular innovation
1. Mechanisms
Behind every successful transformation lies a choreography of molecular events (bond breaking, bond making, electron shifts, fleeting intermediates) that unfold on timescales too swift for the naked eye but critical to the fate of the reaction.
Our research embraces this invisible theater, seeking not only to identify the key players but to understand their roles, timing, and interactions.
Rather than treating mechanisms as static maps, we view them as dynamic systems shaped by subtle energetic preferences and environmental cues.
To decode these pathways, we combine kinetic profiling, isotope tracing, crystallography and theoretical modeling (to name a few techniques) with a curiosity-driven mindset that favors questions as much as answers.
This mechanistic lens allows us to move beyond empirical optimization toward predictive control, where reactivity is sculpted with intention and selectivity emerges from design rather than chance.
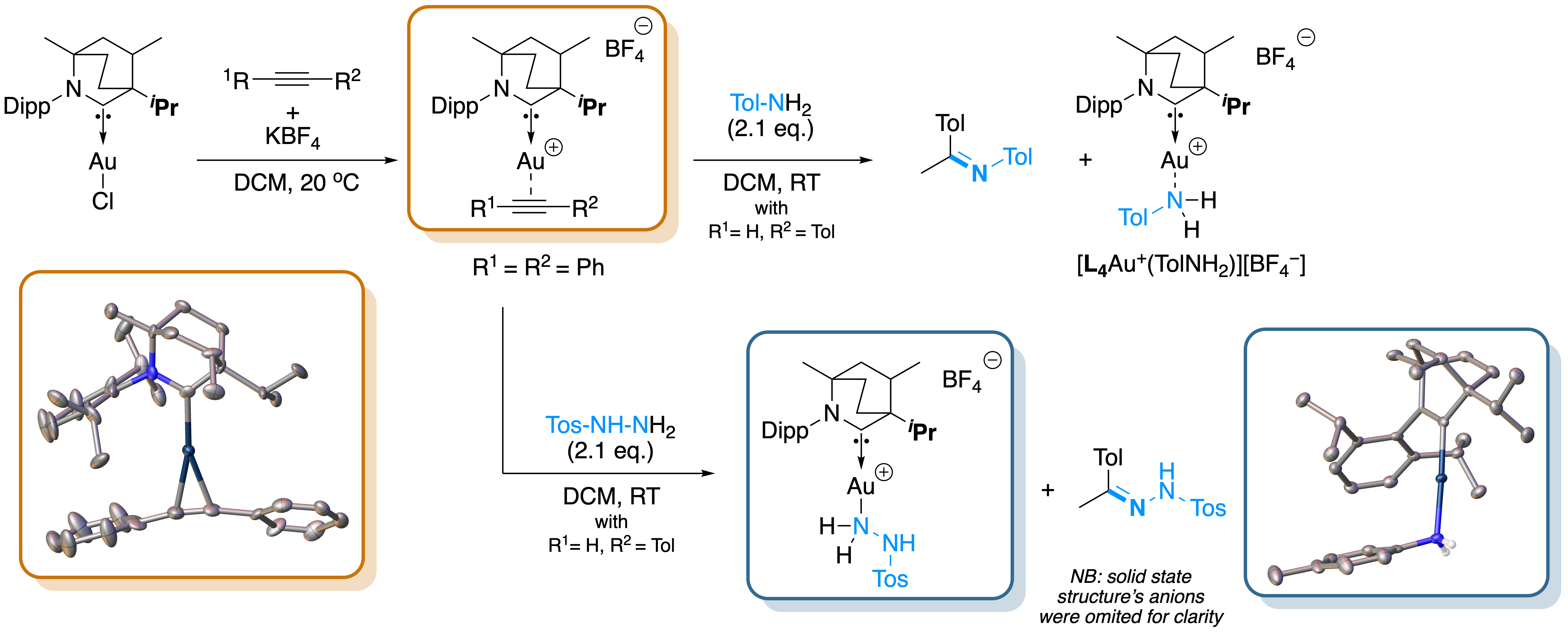
This figure illustrates a mechanistic study of a gold-catalyzed hydroamination and hydrazination reaction
in which key intermediates of the catalytic cycle were isolated. The study provided insight into the exceptional activity of this family of carbene-stabilized
gold catalysts, achieving turnover numbers (TONs) of up to 16,000 and 36,000, respectively.
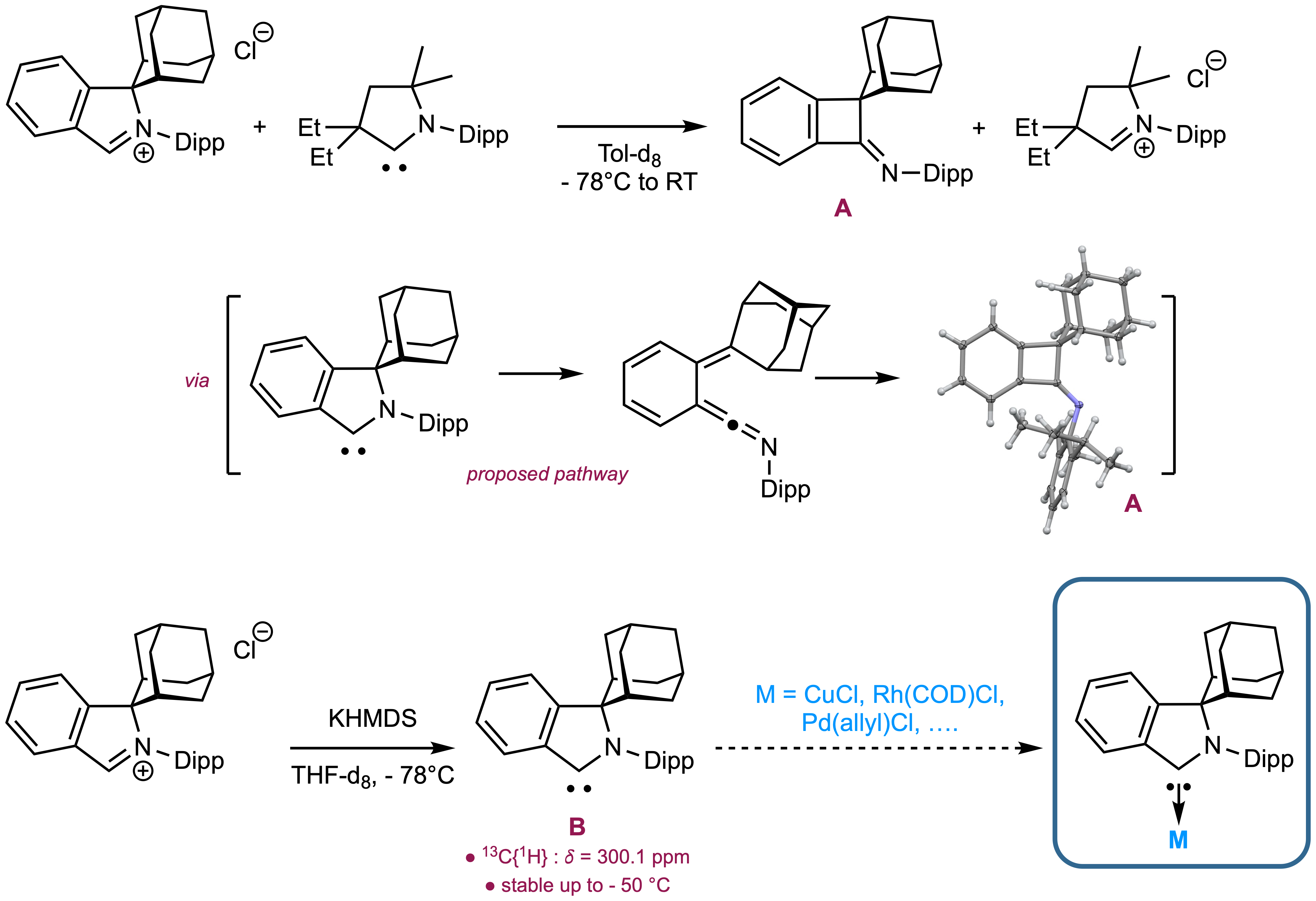
Our group investigates the structural foundations of chemical reactivity in main group compounds, with a particular interest towards carbon allotropes.
Once seen as limited to structural roles, main group elements are now recognized as powerful platforms for reactivity and catalysis, offering sustainable alternatives to transition metals.
Carbenes, exemplify this shift: as ligands or reactive species,
they allow precise modulation of electronic and steric environments and enable access to unusual bonding scenarios. Of specific interest, we aim to explore the design and reactivity of
chiral main group elements, uncovering new pathways for asymmetric catalysis and stereoselective transformations.
Schematic representation of a modular ligand system.
In our work, we are interested in approaching material design as a means of enabling chemical expression at
the molecular level. This is the case with metal hydrides−compounds featuring a direct bond between
a metal and a hydrogen atom ("M−H")−which are fundamental to many catalytic and materials processes.
Notably, they serve as key intermediates in hydrogenation, hydrofunctionalization, and reductive coupling reactions,
and their behavior is central to both homogeneous and heterogeneous catalysis. While noble metal hydrides
are well-studied, the hydrides of earth-abundant metals remain comparatively underexplored despite their
potential to offer cost-effective, sustainable alternatives. These systems often exhibit non-classical
bonding and reactivity, providing an ideal testing ground for advancing our understanding of structure–reactivity
relationships. By studying abundant metal hydrides in well-defined molecular settings, we seek to uncover principles
that can inform catalyst design, surface chemistry, and industrial processes, particularly those related to energy
storage and green synthesis.
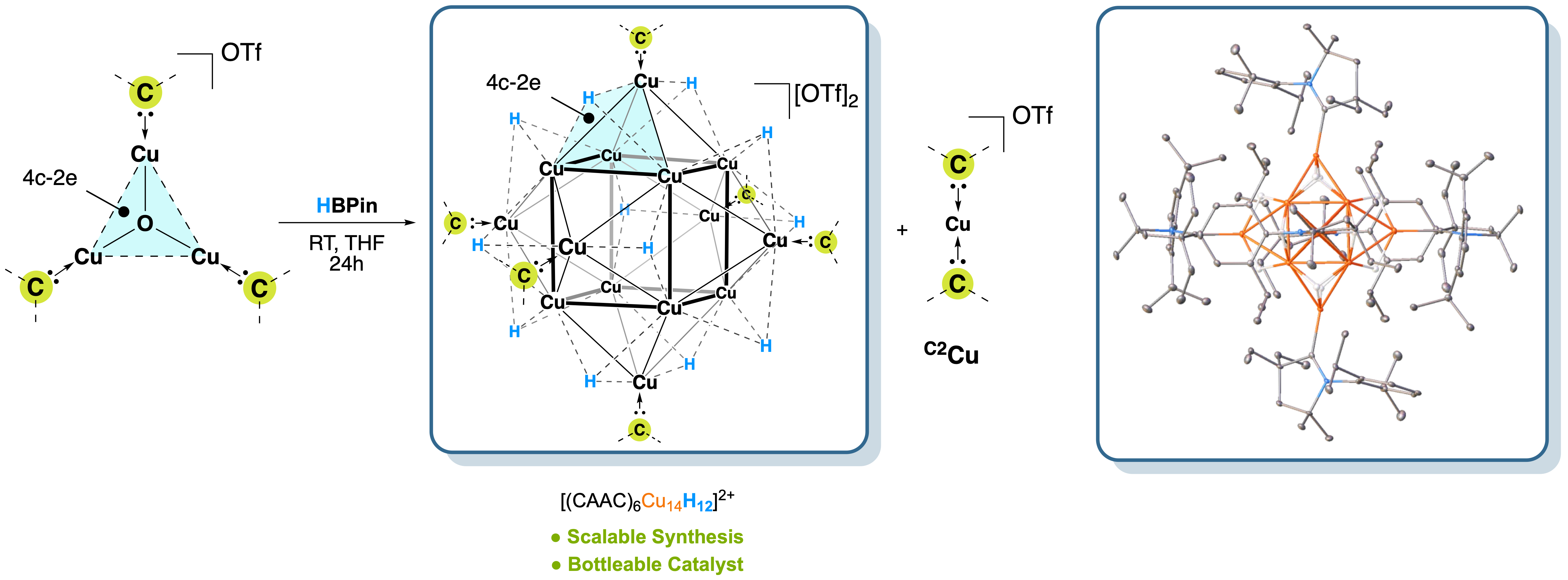
This figure illustrates the structure of a carbene-stabilized
copper hydride cluster, shedding light on the bonding and reactivity of earth-abundant metal hydrides. Our study reveals
how carefully designed carbene ligands can stabilize multimetallic Cu–H assemblies, offering new insights into hydrogen
transfer, metal–metal cooperativity, and potential models for heterogeneous surface reactivity.
Although our primary focus lies in the design and mechanistic understanding of reactive molecular systems, many of these architectures naturally lend themselves to catalytic applications. We actively explore such opportunities in collaboration with leading catalysis groups, including the
Engle Lab at Scripps and the
Mauduit Lab at ENSCR, whose complementary expertise enables robust evaluation and deployment of our compounds in synthetically relevant transformations. These partnerships allow us to assess the translational potential of our systems under rigorous conditions, while contributing molecular design, mechanistic rationale, and reactivity profiling. This collaborative strategy strengthens the broader impact of our work and underscores the relevance of fundamental discovery in addressing catalytic challenges.
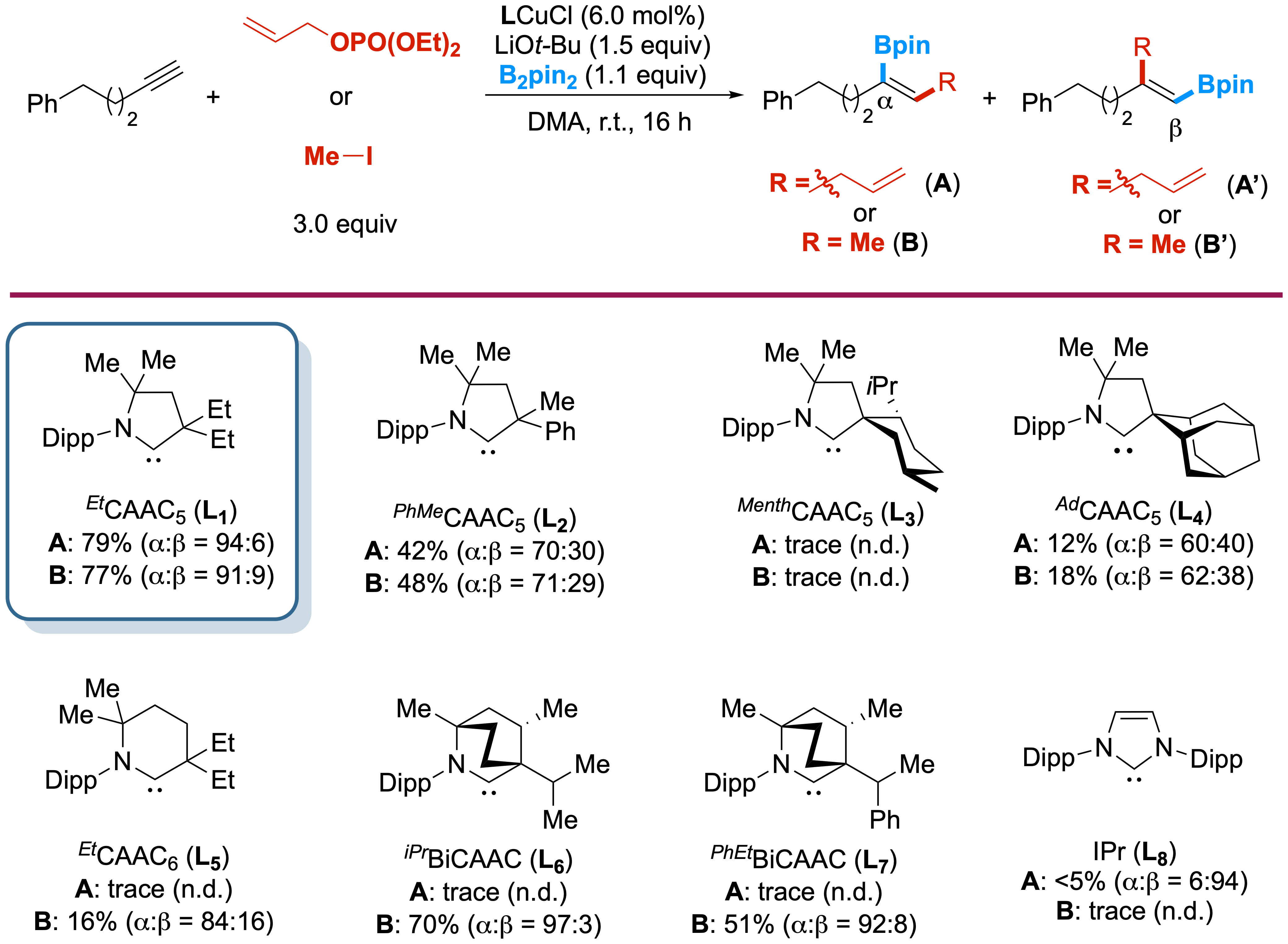
In this figure (with the Engle group), we explored how small changes to the
ligand (this part of the catalyst that surrounds and fine-tunes the metal center) can dramatically affect the outcome
of a chemical reaction. By testing a library of copper catalysts supported by different carbene ligands, we identified
a lead structure that promotes high efficiency and strong control over product selectivity. This study highlights how
careful molecular design helps improve the performance and precision of chemical transformations.
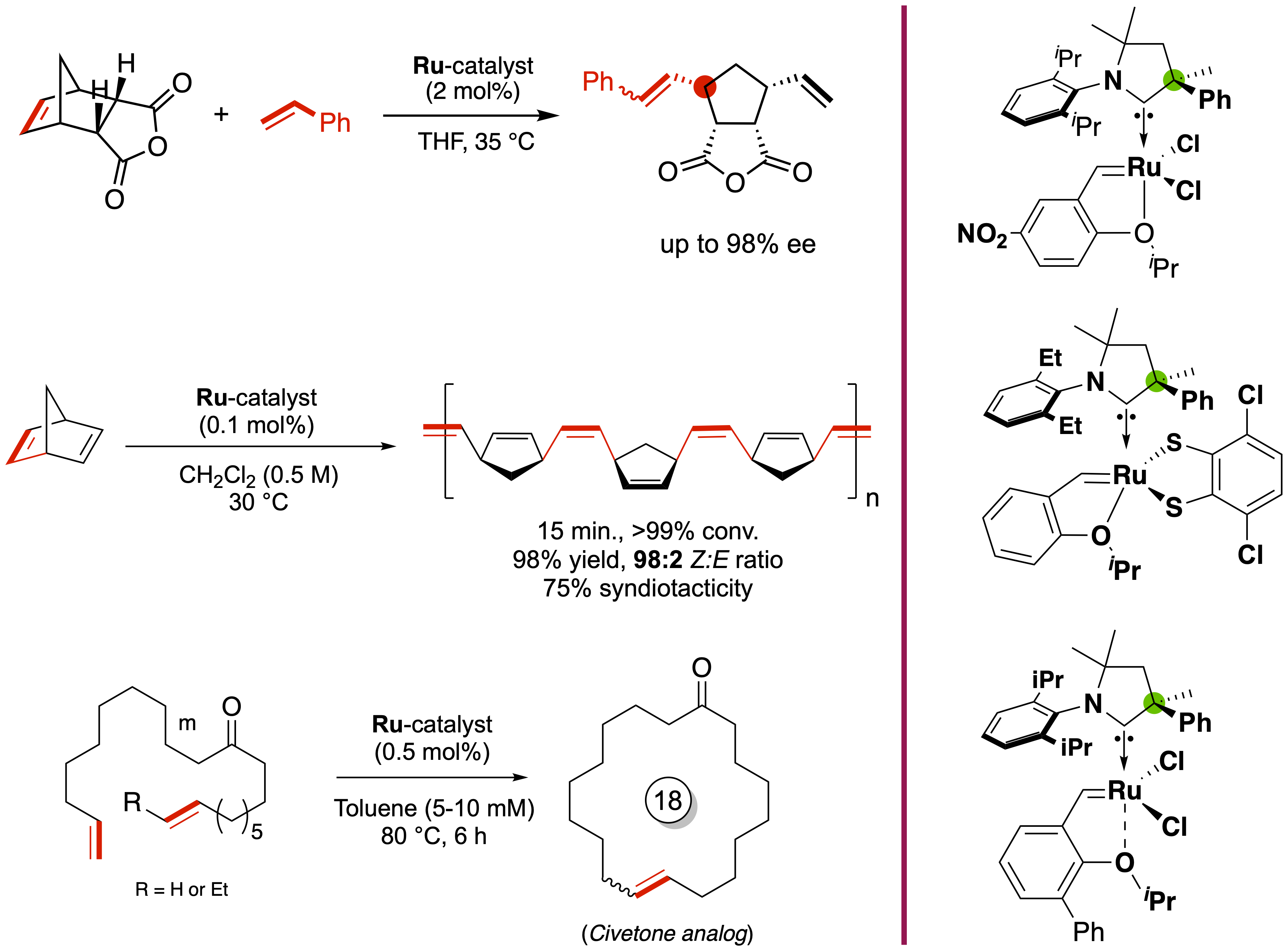
In this figure (with the Mauduit group), we highlight
the catalytic activity of enantiopure olefin metathesis catalysts that precisely
control the "handedness" (chirality) of chemical reactions. This level of control is crucial in pharmaceutical synthesis,
where many drugs exist as mirror-image molecules: only one of which may be therapeutically effective, while the other can be
inactive or even harmful. By enabling the selective formation of the desired enantiomer, chiral catalysts contribute to safer,
more efficient drug development and synthesis.
While we are certainly not specialists in theoretical chemistry, our group actively uses density functional theory (DFT) and
related computational approaches to complement our experimental efforts. These tools help us probe electronic structures, map plausible reaction pathways,
and better understand the steric and electronic features of ligands and catalysts. We are also fortunate to work with experts in the field (
Macgregor Lab,
ITEM Lab ) who help us rationalize
observed reactivity trends and to refine mechanistic proposals. This computational perspective has proven invaluable in guiding experimental design and
interpreting complex chemical behavior. Ultimately, our goal is to integrate theory and synthesis in a pragmatic, question-driven manner that deepens insight
into molecular reactivity and catalysis.
Two short clips illustrating how computational chemistry enables visualization of electronic structures and,
in this case, reveals the origins of differing reactivities in metal hydride clusters (with ITEM Lab).